








 首頁
首頁【專欄】解碼罕見疾病:從全球挑戰到臺灣的機會

我們是否曾經關注過罕見疾病的基礎科學研究?
你可能聽過癌症、糖尿病或心血管疾病,但你是否知道,全世界有超過三億人,正與一種或多種「罕見疾病」奮戰?這些疾病發生率極低,往往每萬人中不到一人罹患,但對患者與其家庭而言,卻可能是漫長且充滿未知的醫療旅程。面對這些複雜、罕見且多數無藥可醫的病症,科學家們正在不斷挑戰現有的醫學疆界。
根據估計,目前全球已知的罕見疾病超過 7,000 種,總共影響人數超過三億人,也就是說,平均每 20 人中,就有一人受到罕見疾病的影響。對患者與其家庭而言,這些病不僅是醫療上的挑戰,更是一場漫長且充滿不確定性的生活旅程。他們不只要面對疾病本身帶來的生理負擔,還經常遭遇診斷延誤、治療資源不足、藥物開發困難等問題。
正因為罕病的多樣性與複雜性,全球越來越多的國家投入資源進行相關研究與政策推動。歐盟、美國、日本等地皆設有專門的罕見疾病政策單位,推動基因資料庫、臨床試驗平台與新藥審查機制。聯合國與世界衛生組織也逐漸強調罕見疾病的公共健康重要性。這些國際間的合作展現了共同對這個領域的重視與承諾,也讓病友與家庭感受到更實質的支持。特別是在資料庫建構、跨境臨床資料整合,以及針對極少數患者開發個人化醫療方面,已出現初步成效。
臺灣的罕病現況
於1999年,臺灣民間發起的罕見疾病基金會成立,開始推動醫療人權提倡、病友權益爭取、生育關懷推廣及政策制度的改革,多面向齊頭並進。並以「我們不可能照顧孩子一輩子,但制度可以」的信念,凝聚了病友家庭的力量,深刻撼動社會各界。罕見疾病基金會從初期關懷病友到如今發展出八大宗旨(圖一),試圖建立一完整的罕病病患權益保障網絡。
目前更持續落實病友在地化服務、全人關懷、全方位照護計畫、國際交流合作及罕見疾病防治研究等工作。然而在臺灣的罕病基礎科學研究,仍不及罕病臨床研究或統計研究活絡,這落差需更多基礎科研機構或政府官員在資源分配上有更多同理心,以及更多基礎科研人材的關注、正視及投入。

圖一、罕見疾病基金會從初期關懷病友到如今發展出八大宗旨,試圖建立一完整的罕病病患權益保障網絡(圖片來源:罕見疾病基金會)
臺灣自 2000 年起實施《罕見疾病防治及藥物法》,成為亞洲最早將罕見病納入國家保障體系的國家之一。根據衛福部統計,截至目前臺灣已公告 226 種罕見疾病,納入健保給付者逾 100 種。雖然病友數量不多,但他們同樣需要長期的醫療照護、心理支持與社會接納,特別是面對高額藥費與長期治療所帶來的壓力。
與此同時,少子化成為臺灣長期發展的關鍵挑戰。如何保障下一代健康成為社會政策的優先目標。新生兒篩檢與精準醫療結合,能有效提早發現先天性罕病,避免病情惡化與後續照護成本,因此被視為醫療系統進步的重要指標。世界衛生組織自 2014 年起即鼓勵各國發展新生兒篩檢系統,並結合健康保險制度強化對新生族群的照護。
在眾多罕見疾病中,「溶小體儲積症(Lysosomal Storage Disorders,LSDs)」是一大類代謝不正常病群,目前全球已知超過70種不同的亞型1。LSD 主要是由於細胞內的「溶小體(lysosome)」缺乏特定酵素,無法正常水解代謝物,而導致廢棄代謝物堆積,進而引起細胞功能失調。這些疾病多數從兒童期開始發病,且進展快速,症狀可能包括肝脾腫大、骨骼異常、神經退化,甚至影響視力與智力發展。三分之二的溶小體儲積症會影響中樞神經系統,因此若無法及時診斷與治療,將造成不可逆傷害。
溶小體儲積症可依代謝物類型分為四大類:鞘脂沉積(sphingolipidoses)、黏多醣儲積症(mucopolysaccharidoses)、肝醣儲積症(glycogen storage disorder)與醣蛋白儲積症(glycoproteinoses)。其中黏多醣儲積症與醣蛋白儲積症常出現骨骼異常、器官腫大與神經症狀,臨床表現雖類似,但致病酵素與代謝物卻全然不同。因此突顯準確診斷的重要性,才可能有對症下藥的方式。這些疾病不僅難以在早期被識別,在臨床醫師診斷與用藥決策上,也經常缺乏足夠的量化資料佐證。
臨床診斷與挑戰
目前臨床上主要透過四種指標進行溶小體儲積症診斷包括:(i)酵素活性測定、(ii)代謝物堆積程度分析、(iii)基因定序與(iv)臨床症狀判讀。第一線常使用的為質譜分析測定酵素活性,但受限於合適酵素基質及量化所需內標種類不足,現僅限約為10項,這使得新生兒篩檢的效率與廣度皆受限2、3。
另一方面,代謝物堆積雖能直接反映病情進展與藥效,但因分子複雜性高,如高甘露醣寡醣(high mannose glycans)、醣胺聚醣(glycosamininoglycan)等,皆缺乏臨床適用的簡便定量分析工具。此外,次世代定序(NGS)可提供致病基因資訊,但約有 20~30% 的基因變異屬於「不確定意義變異(VUS)」,若無生化表現佐證,將難以用於臨床決策及用藥前後比較或追蹤成效。最後,臨床症狀的解讀亦需小心,因同一疾病在不同個體表現差異極大,而不同病亦可能有重疊症狀。這也意味著診斷需要結合各種資料來源,相互佐證,才能建立可信的診療判斷。
值得注意,上述前兩項為重要生化基礎科研範疇,仍有很多待開發及探索的空間。在新生兒罕病篩選中,酵素活性測定為第一關卡,所以需要快速且大通量的方便檢測方式。最主要目的是將大部份的正常人去除,留下不確性或高風險之個體由其他後續檢測把關。其中代謝物堆積程度分析為較明顯客觀及較易定量之方式。此也為臨床用藥的重要參考指標。因此,如果能針對代謝物堆積定量分析,建立全世界統一規範,將有助於地域及人種之分析及預測。
為解決目前診斷與追蹤困難,我們的研究團隊提出以溶小體儲積症為切入點,整合化學合成及分析、生化、和化學生物學資源,開發新一代臨床檢測工具,建立「以需求為導向」的研究模式。研究重點包括:(1)開發新型醣相關水解酵素之基質與內標分子,期待對酵素活性測定的深度及廣度有明顯覆蓋率,並建立同時多工質譜篩檢平台,(2)針對具挑戰性的溶小體儲積症建立方便且定量的代謝物堆積分析方法,並找出具臨床價值的診斷指標。
其中在針對 α-甘露醣儲積症的研究中,團隊成功建立了世界第一個可定量溶小體甘露醣堆積的臨床診斷檢測平台,並已申請專利,亦獲合作者馬偕醫院納入重點發展項目。這項成果展現從基礎研究到臨床實務轉譯的可能,也證明臺灣具備發展全球罕病代謝檢測中心的潛力。透過整合跨領域技術與臨床需求,研究團隊開啟了一個由實驗室研發走向臨床需求的成功案例,為罕病檢測建立了台灣模式。
臨床醫療與挑戰
目前溶小體儲積症的主要治療方法有:酵素替代療法(enzyme replacement therapy,ERT)、基質減少療法(substrate reduction therapy,SRT)、造血幹細胞移植(HSCT)、藥理伴護小分子療法(Pharmacological chaperone)、基因治療等幾大類4。另外,mRNA療法也在研發中。然而每種都有不同的機制和給藥策略,也有各自治療方法對不同溶小體儲積症的限制。舉例,最常見及常用的是「酵素替代療法」(Enzyme Replacement Therapy,ERT),也就是定期給予病人缺乏的酵素,以協助分解體內堆積的物質。然而這類療法往往費用高昂,且無法有效穿透血腦障壁,因此對於中樞神經系統受影響的疾病效果有限。此外也有針對特定病症的生合成路徑而開發的抑制劑,以期待減緩代謝物堆積量。上述研發皆需要厚實的基礎科研人才來支持。鄭老師團隊不僅有研究小分子抑制劑以應用於SRT,另外也正在開發「藥理伴護小分子」,提供更多元、個別化的治療可能性。
小分子藥物的潛力與未來
我們的研究團隊近年來就致力於設計與開發這樣的小分子,用以修復某些罕見病患者體內失能的酵素。藉由天然物化學結構的啟發同時結合組合式化學的合成技術平台(natural product-inspired combinatorial chemistry,NPICC),研究團隊過去以苦馬豆素為骨架,重新合成打造出約400個結構變易性大且特殊之類天然物分子群,並快速逐一進行相對酵素抑制活性篩選,再利用電腦輔助進一步優化分子結構,得到具有高度專一性之高基氏體甘露醣水解酶抑制劑ACK900,並且對於多株癌細胞具有抑制效果(圖二)5。
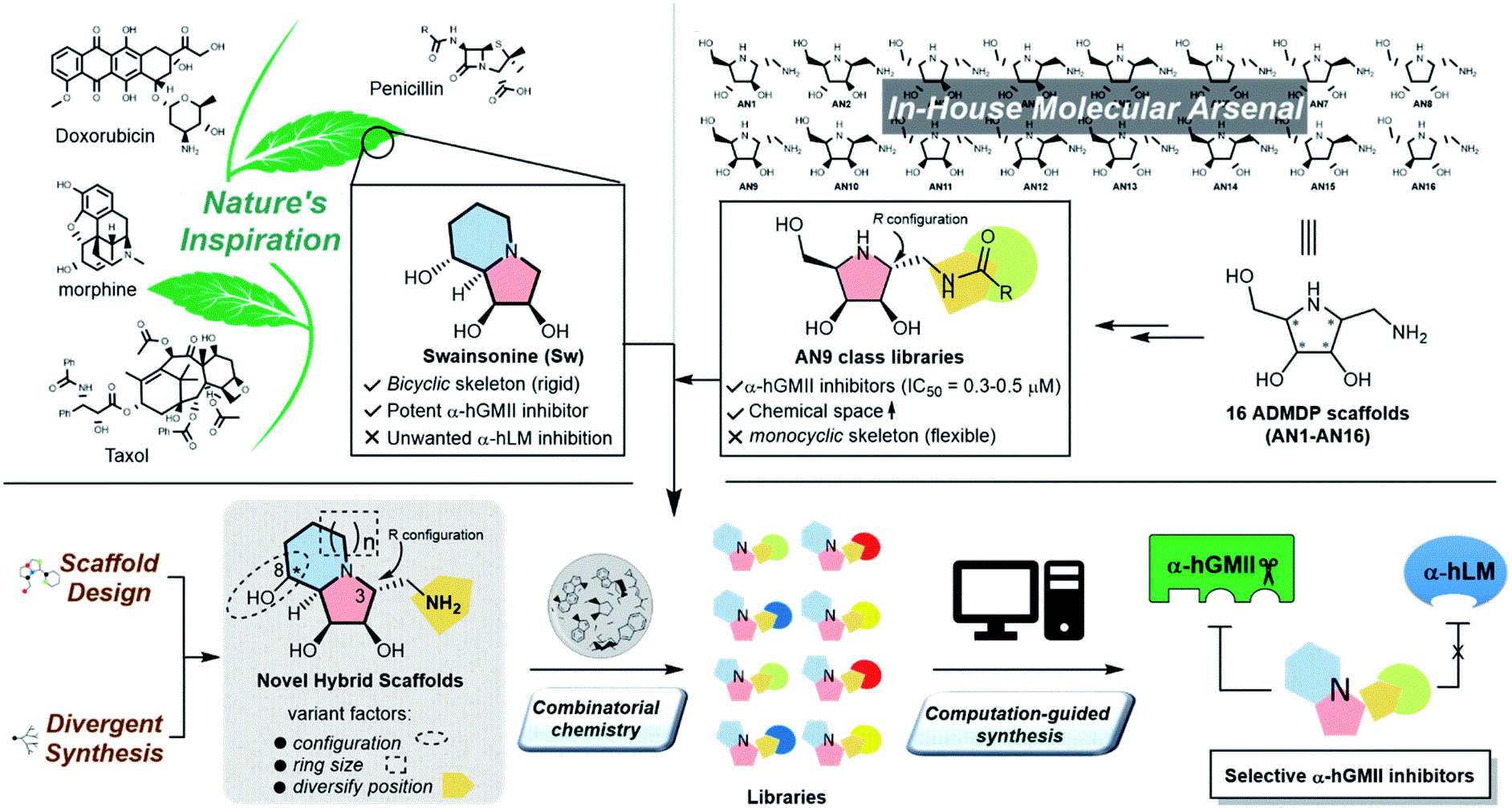
圖二:研究團隊過去以苦馬豆素為骨架,重新合成打造出約400個結構變易性大且特殊之類天然物分子群,並快速逐一進行相對酵素抑制活性篩選,再利用電腦輔助進一步優化分子結構,得到具有高度專一性之高基氏體甘露醣水解酶抑制劑ACK900,並且對於多株癌細胞具有抑制效果。
什麼是「藥理伴護小分子」?這些化合物可以想像成是幫助病態蛋白質恢復穩定與功能的「輔導老師」,當蛋白質因突變而摺疊鬆散錯誤、失去活性時,這類分子能穩定其結構,使其能正確運作。利用「由天然物所啟發之組合式化學」的技術平台,鄭老師的研究團隊成功開發出能夠對於法布瑞氏症、龐貝氏症、高雪氏症和黏多醣症第一型(MPSI)等罕病具有治療潛力的酵素穩定分子來穩定特定蛋白質大分子藥物或藥理伴護小分子來穩定細胞內特定突變的蛋白質,並也發表了多篇國際期刊以及多項專利6、7、8、9。這樣的化學策略不僅提供了新穎的治療潛力,也為傳統治療方式(如酵素替代療法)提供輔助可能。
最近,研究團隊發現五環亞胺醣ACK170可與半乳醣水解酶在中性pH值環境下形成強力靜電交互作用。而這個作用力會在pH值酸性的環境中因為羧基的質子化而減弱(圖三)。結果證明在亞胺醣分子上引入一個額外的胺基,可提升藥理伴護小分子在pH中性環境中對於酵素的結合力和pH值選擇性,對於未來開發新型藥理伴護小分子奠定新的里程碑6。
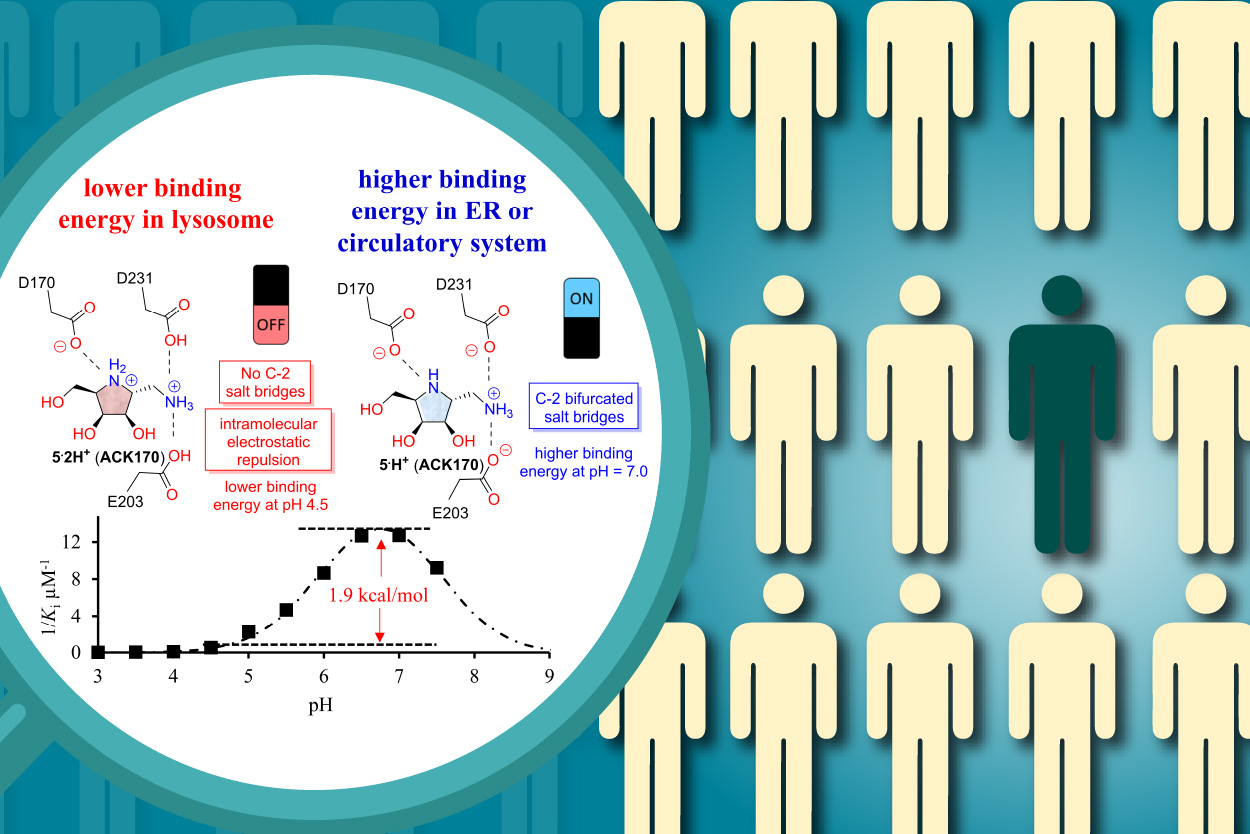
圖三:研究團隊發現五環亞胺醣ACK170可與半乳醣水解酶在中性pH值環境下形成強力靜電交互作用。而這個作用力會在pH值酸性的環境中因為羧基的質子化而減弱。
相較於酵素替代療法與基因療法,小分子藥物具備分子量小、可口服與成本相對低廉等優勢,能補足現有治療的限制。鄭老師的團隊也持續開發能穩定錯誤摺疊酵素的小分子伴護劑,讓酵素能夠順利進入溶小體並發揮功能。這些化合物不僅可用於治療,亦可作為生化診斷工具的延伸。若能成功推進至臨床,將大幅降低患者每年上百萬甚至上千萬的藥物支出,讓罕病治療更具可近性。展望未來,精準醫療將是罕病診治的核心方向,結合高通量診斷平台、個別化治療策略與小分子工具開發,讓病患不再受限於單一標準流程。
臺灣在臨床罕病的角色及機會
臺灣有令各國稱羡的全民健保制度,也有地理區域及人種分佈的代表性及特色。若能持續強化基礎化學及科學與臨床應用之連結,不僅能服務在地病患,更有機會成為全球罕病研究與驗證的重要據點。透過深耕技術、跨界合作及以病患為中心的思維,我們不僅能提升科學創新價值,更能實現罕病院床轉譯的成果,期打造一個更完善的制度及一個真正公平而有溫度的醫療環境。罕病雖罕見,卻不該被忽視。正是這些疾病,提醒我們醫療不該遺落任何一人;未來希望能有基礎科研研究者加入,讓我們看見,科學與社會關懷是可以並行不悖的。
致謝
感謝李皇毅博士共同整理文稿資料。感謝慈濟大學分子生物暨人類遺傳學系教授及台灣尤塞氏症暨視聽弱協會理事長靖永皓博士的內容討論。感謝罕見疾病基金會陳冠如執行長的熱心討論並由基金會同意使用圖一。
參考文獻
(1) Platt, F. M.; d’Azzo, A.; Davidson, B. L.; Neufeld, E. F.; Tifft, C. J. Lysosomal storage diseases. Nat. Rev. Dis. 2018, 4, 27.
(2) Gelb, M. H. Newborn Screening for Lysosomal Storage Diseases: Methodologies, Screen Positive Rates, Normalization of Datasets, Second-Tier Tests, and Post-Analysis Tools. Int. J. Neonatal Screen 2018, 4.
(3) Pančík, F.; Pakanová, Z.; Květoň, F.; Baráth, P. Diagnostics of lysosomal storage diseases by mass spectrometry: a review. Chem. pap. 2022, 76, 3995-4004.
(4) Beck, M. Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol. 2018, 60, 13-18.
(5) Chen, W.-A.; Chen, Y.-H.; Hsieh, C.-Y.; Hung, P.-F.; Chen, C.-W.; Chen, C.-H.; Lin, J.-L.; Cheng, T.-J. R.; Hsu, T.-L.; Wu, Y.-T.; et al. Harnessing natural-product-inspired combinatorial chemistry and computation-guided synthesis to develop N-glycan modulators as anticancer agents. Chem. Sci. 2022, 13, 6233-6243.
(6) Li, H.-Y.; Lin, H.-Y.; Chang, S.-K.; Chiu, Y.-T.; Hou, C.-C.; Ko, T.-P.; Huang, K.-F.; Niu, D.-M.; Cheng, W.-C. Mechanistic Insights into Dibasic Iminosugars as pH-Selective Pharmacological Chaperones to Stabilize Human α-Galactosidase. JACS Au 2024, 4, 908-918.
(7) Li, H.-Y.; Chen, W.-A.; Lin, H.-Y.; Tsai, C.-W.; Chiu, Y.-T.; Yun, W.-Y.; Lee, N.-C.; Chien, Y.-H.; Hwu, W.-L.; Cheng, W.-C. A practical synthesis of nitrone-derived C5a-functionalized isofagomines as protein stabilizers to treat Gaucher disease. Commun. Chem.2024, 7, 91.
(8) Lin, H.-Y.; Chang, S.-Y.; Teng, H.-H.; Wu, H.-J.; Li, H.-Y.; Cheng, C.-C.; Chuang, C.-K.; Lin, H.-Y.; Lin, S.-P.; Cheng, W.-C. Discovery of small-molecule protein stabilizers toward exogenous alpha-L-iduronidase to reduce the accumulated heparan sulfate in mucopolysaccharidosis type I cells. Eur. J. Med. Chem. 2023, 247, 115005.
(9) Li, H.-Y.; Lee, N.-C.; Chiu, Y.-T.; Chang, Y.-W.; Lin, C.-C.; Chou, C.-L.; Chien, Y.-H.; Hwu, W.-L.; Cheng, W.-C. Harnessing polyhydroxylated pyrrolidines as a stabilizer of acid alpha-glucosidase (GAA) to enhance the efficacy of enzyme replacement therapy in Pompe disease. Biorg. Med. Chem. 2023, 78, 117129.
