研究之中,找到我們是誰 | 中央研究院(2025年版形象影片)
空拍
院區風景
圖書館
南部院區
聲明專區
中研院學術大會
中央研究院第36次院士會議今(6)日下午舉行專題討論,邀請國家科學及技術委員會吳誠文主任委員與教育部鄭英耀部長,分別以「開創國家科研發展新局」及「鏈結全球、布局未來:臺灣新世代人才培育與國際共創」為題,聚焦臺灣科研政策與人才戰略的政策方向,與院士相互交流。
中央研究院第36次院士會議今(6)日邀請中研院名譽院士、美國國家工程學院(National Academy of Engineering, NAE)前院長、伊利諾理工學院前校長約翰.安德森(John L. Anderson),以及中研院院士、美國國家工程學院院長劉金智潔(Tsu-Jae King Liu)發表主題演講,並由本院廖俊智前院長與周美吟副院長共同與談。
中央研究院第36次院士會議今(6)日正式開幕,超過200位海內外院士齊聚臺灣,共同關心國家學術發展與科研未來。為期4天的會議舉行院士證章頒發、主題演講、專題討論等,並將選出第35屆院士暨名譽院士。陳建仁院長於開幕典禮致詞表示,本次院士會議是他接任院長後首次主持的大型學術盛會。中研院即將迎來創院百年,站在邁向第二個百年的重要起點,期盼與全體院士及同仁攜手並進,以智慧推動頂尖研究,以勇氣帶動創新突破,以慈悲帶來人文關懷,使中研院持續發揮學術引領角色,成為亞太卓越研究重鎮,並透過知識創新回應國家與全球面臨的重要挑戰。
中央研究院將於今(6)日至9日舉行第36次院士會議。依循歷屆傳統,昨(5)日晚間以「仲夏夜音樂會」揭開院士會議序幕,邀請國內外院士、貴賓及院內同仁齊聚,以音樂交流情誼。除了享有盛譽的中央研究院合唱團擔綱演出,本次更有多位院士登臺獻藝,展現科學家的人文風采,與現場來賓共度知性的仲夏夜饗宴。
中央研究院陳建仁院長昨(2)日出席本院社會學研究所「學術諮詢委員會議暨30週年慶」活動,會中邀請美國杜克大學社會學系榮休教授林南院士、美國普林斯頓大學社會學及國際研究講席教授謝宇院士,以及多位學術諮詢委員與會,回顧並展望社會學研究與國際交流的發展方向。
中央研究院陳建仁院長近日接見蒙古科學院生物研究所(Institute of Biology, Mongolian Academy of Sciences)Gantulga Davaakhuu所長訪團,雙方就公共衛生政策、病毒性肝炎防治、生物醫學研究及人才培育議題深入交流。本院生物化學研究所、生物多樣性研究中心、國立宜蘭大學生物資源學院,與蒙古科學院生物研究所四方也共同簽署合作備忘錄,建立長期合作機制。陳院長表示,中研院長期重視國際合作,此次攜手國立宜蘭大學,結合研究機構與大學的科研量能,展現跨界資源整合的成果,並象徵臺蒙科學合作邁向新里程碑,共同推動研究計畫、人員互訪及人才培育。
行政院公共工程委員會陳金德主任委員今(1)日率團拜會中央研究院陳建仁院長,雙方就宜蘭科研布局及重大建設交換意見,期盼透過跨部會合作,完善科研基礎建設,提升臺灣整體科研競爭力,並促進區域均衡發展。
由中央研究院協辦、國立自然科學博物館(下稱科博館)主辦的「看見典範:臺灣科研創新與成就」特展今(1)日盛大開幕,帶領民眾走近12位中研院院士的科研人生。陳建仁院長表示,感謝科博館策劃此次展覽,透過深入淺出的方式介紹不同科學背景院士的學思歷程,讓社會大眾認識科學家的工作如何探索未知,回應自然、健康與環境等人類共同面臨的挑戰。
第75屆林島諾貝爾獎得主會議將於2026年6月28日至7月3日在德國林島舉行。來自88個國家、逾630位年輕科學家獲邀,與約75位諾貝爾獎得主面對面交流。本院物理研究所柯書華(Shuvadeep Karmakar)博士與應用科學研究中心黃郁涵博士,2位優秀的博士後研究學者通過嚴格的國際遴選,代表臺灣與會。
中央研究院今(22)日舉行「新、卸任副院長歡迎、歡送茶會」,由陳建仁院長主持,宣布原子與分子科學研究所特聘研究員周美吟院士、分子生物研究所特聘研究員蔡宜芳院士,以及經濟研究所特聘研究員彭信坤院士,擔任本院副院長;原副院長唐堂院士卸任。
中央研究院今(18)日舉行院長交接典禮,由陳建仁院士自廖俊智院長手中接下本院第13任院長之職務。蕭美琴副總統蒞臨監交,蔡英文前總統受邀出席,李遠哲前院長、本院院士、院內同仁及各界學研貴賓共同見證中研院承先啟後的重要時刻。
2026年中央研究院「中研講堂」科普講座前進高雄鳳山高中,從AI、永續農業到民主自由,探討「面對未來的提問與選擇」。這場結合自然科學與人文思辨的講座,吸引逾500名師生與民眾參與,座無虛席。現場同學們踴躍的提問,與3位講者互動,相當精彩。
國際事務處
2026/07/03
專題演講「AI如何學會思考?從大腦到人工智慧的探索」
人事室
2026/06/25
本院語言學研究所所長由研究員曾淑娟博士接任,陳院長主持交接典禮。
學術及儀器事務處
2026/06/23
115年度第2梯次「國內學人短期來院訪問研究」核定通過名單
2026/06/17
中央研究院與阿曼國立科技大學簽署合作備忘錄 深化學術與研究合作
近代史研究所
2026/06/16
近史所檔案館開放中西醫書及醫案 推動醫療公衛史研究
原子與分子科學研究所
2026/06/15
徵件〉116年度本院「材料與分析科技探索計畫」,即日起受理申請
2026/06/10
本院115年模範公務人員暨工作績優人員獲選名單與個人優良事蹟表
2026/06/01
中央研究院115年度第一次醫學研究倫理教育訓練
2026/05/25
116年度「國立臺灣大學與中央研究院創新性合作計畫」即日起至115年7月10日止受理申請
2026/05/22
近代史研究所陳冠任助研究員獲選英國皇家歷史學會會士
2026/05/21
115年度「中央研究院人文社會科學博士候選人培育計畫核定名單」
2026/05/15
2026-2027年度本院與波蘭科學院共同徵求國際合作計畫核定公告
秘書處
2026/05/13
中研院藝文活動〉高雄市管樂團:《跨樂一甲子》流行音樂會
2026年中央研究院藝文活動表演資訊
2026/05/12
116年度「臺北、臺中、高雄榮民總醫院、三軍總醫院與中央研究院合作研究計畫」,自115年6月1日起至115年7月15日止受理申請
2026/05/08
2026年「第34屆新科院士演講」系列活動訂5月15日至9月29日舉行
2026/05/04
116年度本院新增任務導向生技研究計畫及因應流行病研究計畫,即日起受理申請
2026/05/01
本院即日起至115年6月12日止受理志願服務人員報名
社會學研究所
2026/04/30
研究調查〉「臺灣社會變遷基本調查計畫」第九期第二次正式面訪調查
2026/04/27
活動報名〉2026年知識饗宴 —朱家驊院長科普講座「量子科技與奈米科技對於現代文明的發展」
中央研究院「人文社會科學博士生菁英獎學金」自115年5月15日起至6月15日止受理線上申請,逾期恕不受理。
2026/04/16
「臺灣橋梁計畫」諾貝爾獎得主系列講座「基礎科學之國際合作:實務 經驗分享」
2026/04/15
本院地球科學研究所所長由特聘研究員馬國鳳博士接任,廖院長主持交接典禮。
2026/04/14
本院職場友善辦公室成立 打造安心溝通新管道
基因體研究中心
2026/04/10
陳建仁院士榮獲2026年亞太肝臟研究學會(APASL)Okuda-Omata傑出奬
2026/04/08
曾志朗院士於第14屆華人心理學家學術研討會獲頒2026年「終身成就獎」
2026/03/26
「臺灣橋梁計畫」諾貝爾獎得主系列講座「生命分子、人工智慧與人類 健康」
2026/03/24
有學有術,鑑往知來:沙崙會議揭開百年院慶序幕
永續科學中心
2026/03/18
活動後記〉2026淨零城市展:中研院淨零及AI科研落地,能源大師Amory Lovins重磅開講!
2026/03/12
「臺灣橋梁計畫」諾貝爾獎得主系列講座「大腦的 GPS:我們如何知道 自己身在何處」
2026/03/02
活動報名〉2026年知識饗宴—王世杰院長科普講座 「當臺灣人遇上中國人與美國人:我們如何看待這段微妙的三角關係?」
關鍵議題研究中心
2026/02/26
本院關鍵中心成立「下世代高效太陽能電池技術合作聯盟」
2026/02/25
116年度本院新增前瞻計畫、深耕研究計畫、主題研究計畫、關鍵突破研究計畫即日起受理申請
2026/02/13
活動報名〉「臺灣橋梁計畫」諾貝爾獎得主系列講座「抗體革命」
本院歷史語言研究所所長由研究員陳正國博士接任,廖院長主持交接典禮。
2026/02/11
活動報名〉2026年知識饗宴—錢思亮院長科普講座 「施肥不施肥?—植物的糧食、人類的糧食與環境的永續」
2026/02/10
研究調查〉「臺灣社會變遷基本調查計畫」第九期第二次預試面訪調查
2026/02/03
活動報名〉「臺灣橋梁計畫」諾貝爾獎得主系列講座「什麼是生命?」
民族學研究所
期刊出版〉《民族學研究所資料彙編》第31期
2026/01/30
活動報名〉「中央研究院講座」2025年諾貝爾物理學獎得主演講「量子運算的產業轉型:從科研邁向產業」
2026/01/29
活動報名〉「臺灣橋梁計畫」諾貝爾獎得主系列講座「為什麼你應該喜愛基因改造食品」
化學研究所
化學所陳玉如特聘研究員榮獲HUPO「傑出蛋白質體學成就獎」
出版中心
2026/01/20
展覽訊息〉2026台北國際書展:「藍圖(Blueprint)」主題特展
2026/01/19
活動報名〉「臺灣橋梁計畫」諾貝爾獎得主系列講座「是否存在「B行星 」?—人類會移居到系外行星嗎?」
2026/01/09
活動報名〉「臺灣橋梁計畫」諾貝爾獎得主系列講座「雷射與量子物理 」
2026/01/05
研究調查〉「臺灣社會變遷基本調查計畫」第九期第一次正式面訪調查(延長公告)
2025/12/29
115年度「人文社會科學博士候選人培育計畫」自即日起受理申請
2025/12/23
活動報名〉2026年知識饗宴—蔡元培院長科普講座 「你與人工智慧:與科技共進」
生物多樣性研究中心
2025/12/22
臺灣生物多樣性資訊機構 (TaiBIF) 榮獲2025年GBIF「全球生物多樣性資訊節點獎」 肯定臺灣全球生物多樣性資料共享之貢獻
書籍出版〉《知識的幾何與光影:中央研究院建築選》
2025/12/18
國科會114年度傑出特約研究員本院獲獎名單
2025/12/10
預告修正「臺灣人體生物資料庫資料及檢體使用收費標準」
2025/12/08
活動後記〉「台灣海洋碳移除方法工作坊」圓滿成功
2025/12/03
本院分子生物研究所陳詩允助研究員獲選為歐洲分子生物組織年輕研究學者(EMBO Young Investigator)
2025/11/21
近史所與國防大學簽署學術交流備忘錄 促進戰爭與軍事史合作
活動報名〉「臺灣橋梁計畫」諾貝爾獎得主系列講座「誰能推動全球衝突相關性暴力(CRSV)的終結?— 個人故事的力量與行動主義的角色 」
活動報名〉2025年知識饗宴—胡適院長科普講座 「觸受之眼:VR電影的感官邂逅」
2025/11/18
活動後記〉「2025 AI 引領永續社會國際學術研討會暨政策實務論壇」圓滿成功
2025/11/05
活動報名〉「臺灣橋梁計畫」諾貝爾獎得主系列講座「疾病的終結?—生物醫學的非凡發展及其對人類的意涵」
2025/11/01
本院115年度「國際研究生學程(TIGP)」開始受理申請
2025/10/29
活動報名〉2025年度中央研究院講座 「黑洞:跨越學科的交會點」
2025/10/28
本院生醫轉譯研究中心主任由研究員陳建璋先生接任,廖院長主持交接典禮。
2025/10/23
中研院菁英博士生快速入學試行計畫 助青年學子延續科研之路
2025/10/08
再赴法蘭克福書展 中研院以多元學術視野開啟全球交流對話
2025/09/30
中研院自製量子計算測試平台開放申請使用
2025/09/17
原子與分子科學研究所許良彥研究員榮獲美國物理聯合會《化學物理雜誌》2024年度新興研究者最佳論文獎
人文社會科學研究中心
2025/09/16
活動後記〉「2025中研GIS論壇」圓滿成功
2025/09/15
114年度中央研究院人文社會科學博士生菁英獎學金 共計21名同學獲獎
2025/09/02
研究調查〉「網路徵才詐騙:過度自信與數位素養對使用者之影響的比較研究」網路調查
2025/08/27
基因轉殖鼠核心設施_推廣與教育訓練
2025/08/20
研究調查〉「傳播調查資料庫第三期第四次青少年」調查計畫
2025/08/13
活動報名〉2025年知識饗宴—吳大猷院長科普講座 「探索花朵美麗之謎」
2025/08/08
115年度本院新增「淨零科技研發計畫」即日起至114年10月8日止受理申請
2025/08/04
本院114年模範公務人員暨工作績優人員頒獎典禮業於114年7月25日辦理完竣,茲將本年度獲獎人員之優良事蹟刊登本院網頁公開表揚
2025/07/31
研究調查〉家庭動態調查「2025年預試調查」
2025/07/23
活動後記〉本院攜手UCLA、京都大學合辦 三邊生醫研討會
2025/07/22
本院化學研究所陳玉如特聘研究員榮獲財團法人台灣生技醫藥發展基金會「115年學術講座」
語言學研究所
2025/07/21
《語言暨語言學》期刊新任編輯團隊公告
活動後記〉2025臺灣數位世代研究第二屆國際學術研討會
2025/07/17
研究調查〉「民眾對公聽會的參與意願與想法」網路調查
研究調查〉「人工智慧應用於刑事司法審判的可能與顧慮」網路調查
研究調查〉「提升臨終醫療中的病人自主權」網路調查
研究調查〉「投資與理財態度調查」網路調查
2025/07/15
活動訊息〉本院第13任院長遴選座談會
2025/07/14
中央研究院114年度第二次醫學研究倫理教育訓練
2025/07/09
本院共3位研究人員榮獲第23屆有庠科技論文獎
活動後記〉首屆「中研學術大會」圓滿落幕,跨域對話激盪合作火花
2025/07/08
中央研究院2025年國際研究生學程錄取公告
2025/07/03
演講全紀錄〉首屆中研學術大會 廖院長開幕演講
2025/06/24
歡迎參加〉本院自6月30日至7月3日首度舉辦「中研學術大會」
2025/06/23
研究調查〉「臺灣社會變遷基本調查計畫」第八期第五次正式面訪調查補訪
研究調查〉「臺灣青少年成長歷程研究」正式面訪調查
115年度本院「材料與分析科技探索計畫」,即日起受理申請
2025/06/19
114年度第2梯次「國內學人短期來院訪問研究」核定通過名單
2025/06/12
114年度第2梯次本院新增關鍵突破種子計畫核定公告
2025/06/10
本院近代史研究所與國家圖書館舊籍數位化合作締約 近代史文獻共享傳播
2025/06/09
本院近代史研究所陳冠任助研究員獲2024北美海洋史學會約翰.萊曼圖書獎
2025/06/06
114年度「中央研究院人文社會科學博士候選人培育計畫」核定名單
2025/06/02
研究調查〉「傳播調查資料庫第三期第四次」面訪調查
2025/05/29
114年度第2梯次博士後研究學者申請案核定通過名單
2025/05/26
本院114年度「胡適紀念研究講座」開始受理申請
2025/05/23
第13任院長遴選委員會-研究人員及研究技術人員代表選舉 候選人名單公告
2025/05/22
中央研究院「人文社會科學博士生菁英獎學金」自114年6月1日起至7月1日止受理線上申請,逾期恕不受理。
2025/05/15
2025-2026年度本院與波蘭科學院共同徵求國際合作計畫核定公告
2025/05/14
研究調查〉「臺灣社會變遷基本調查計畫」第九期第一次正式面訪調查
2025/05/13
本院即日起至114年6月30日止受理志願服務人員報名
2025/04/29
研究調查〉「美中對臺政策」網路調查
2025/04/28
115年度「臺北、臺中、高雄榮民總醫院、三軍總醫院與中央研究院合作研究計畫」,自114年6月2日起至114年7月15日止受理申請
2025/04/23
中央研究院114年度第二次人文社會科學研究倫理教育訓練
2025/04/22
推動居家照護專業與永續發展—「居家護理師專業及經營管理課程」圓滿舉辦
2025/04/21
活動報名〉2025年知識饗宴—朱家驊院長科普講座 「從黑洞觀看宇宙奧秘」
2025/04/18
115年度本院新增永續科學研究計畫即日起受理申請
第12屆院務會議研究人員代表選舉當選人名單公告
2025/04/16
中研院藝文活動〉茱蒂口琴樂團「口琴說書人」
2025/04/15
研究調查〉「探討衛生所的公共衛生效益」網路調查
2025/04/11
本院人事室主任異動
農生中心高階光學顯微鏡核心設施 超解析暨共軛焦顯微鏡(Zeiss ELYRA 7 with LSM 980)服務說明會 &蔡司新機(Lattice SIM 3)展示會
2025/04/02
本院與EMBO合辦「敘述性履歷」工作坊及課程
2025/03/25
研究調查〉「傳播調查資料庫第三期第四次青少年調查預試計畫」
研究調查〉「臺灣民眾之政治社會態度」網路調查
2025/03/21
本院細胞與個體生物學研究所王漢津助研究員榮獲2024年美國李氏傳統基金會獎助金
2025/03/20
第12屆院務會議研究人員代表選舉公告
本院民族學研究所所長由研究員周玉慧博士接任,廖院長主持交接典禮。
2025/03/17
中研院藝文活動〉微光古樂集「大航海時代的古樂」
2025/03/13
中央研究院114年度第一次人文社會科學研究倫理教育訓練
中央研究院114年度第一次醫學研究倫理教育訓練
Team AS!行政團隊工作效率再升級 全力支援研究
2025/03/10
本院114年度第2梯次「獎勵國內學人短期來院訪問研究」即日起至114年4月18日受理線上申請
2025/03/05
研究調查〉「社群媒體使用」網路調查
2025/03/04
國家科學及技術委員會113年度傑出研究獎本院獲獎名單
2025/02/28
114年「國際研究生學程(TIGP)暑期實習計畫」錄取名單
2025/02/27
活動報名〉2025年知識饗宴—王世杰院長科普講座 「從民調看臺灣民眾的防衛意志」
2024/12/25
中研院新進研究人員交流會 — 鼓勵合作 跨出舒適圈 挑戰新問題
2024/12/19
2025-2026年度本院與捷克科學院共同徵求國際合作計畫核定公告
2024/12/06
2024年「中央研究院年輕學者研究成果獎」訂於12月9日舉行頒獎典禮
2024/12/02
新書出版〉《研之有物:格物窮理!中研院的 25 堂數理科學課》將舉辦新書分享會
政風室
2024/10/17
本院113年度廉能楷模人員與具體事蹟
2024/09/11
本院113年模範公務人員及工作績優人員獲選名單與個人優良事蹟表
2024/05/09
展望2030年 聚焦願景 提出策略 開始行動
2024/04/09
本院即日起至113年5月31日止受理志願服務人員報名
2024/03/29
113年「國際研究生學程(TIGP)暑期實習計畫」錄取名單
歐美研究所
2024/02/20
美國密西根大學2024年「暑期社會研究量化方法課程」開始報名
2024/01/15
國立臺灣大學林麗瓊院士當選世界科學院2024新任院士
2023/09/13
攜手大學共創雙贏 中研院推「中研學者」計畫
院本部
2023/08/22
院長致同仁信:本院博士生獎助金提升方案
Letter from President Liao to Ph.D. Students: Supplemental Stipends for Academia Sinica Ph.D. Students
數理科學組-資訊科學研究所
自然語言處理與情感分析實驗室
截止:2026/08/07
生命科學組-基因體研究中心
林國儀老師實驗室 / Dr. Kuo-I Lin’s Lab
截止:2026/07/31
人文及社會科學組-人文社會科學研究中心
調查研究專題中心調查訪問組
生命科學組-分子生物研究所
郭惠思老師實驗室
調查研究專題中心
數理科學組-統計科學研究所
黃彥棕研究員
政府統計研究群
院本部-智財技轉處
商業開發暨授權組
人文及社會科學組-社會學研究所
鄭雁馨老師
生命科學組-生物醫學科學研究所
Dr.謝小燕(Shieh,Sheau-Yann ) 實驗室
調查研究專題中心謝淑惠研究員研究室
Dr.謝小燕(Shieh,Sheau-Yann)實驗室
截止:2026/08/05
李宗榮老師
人文及社會科學組-民族學研究所
院本部-人事室
截止:2026/07/13
人文及社會科學組-經濟研究所
截止:2026/07/15
江彥生老師
陳恭平特聘研究員
10/19~07/31
10:00 生物多樣性研究中心
「生命之礁:珊瑚的奧秘與生機」特展
01/10~07/12
09:00 歷史語言研究所
[展覽訊息] 親近國寶──大理石梟形小立雕
04/13~08/06
08:00 生物多樣性研究中心
第八屆珊瑚礁生物多樣性與水下生態調查技術研習會 (7/26-8/6,報名至5/31截止)
06/15~07/15
09:00 基因體研究中心
[報名] 2026基因體流行病學研習營 (8/10~14)
07/06~07/09
09:00 秘書處
第36次院士會議
07/09
10:00 近代史研究所
從紙上邊界到地上邊界:中研院近史所檔案館藏滿文字據研究
14:00 政治學研究所
IPSAS系列演講:When Do Outsiders Become Politically Distinct? Rethinking the Insider–Outsider Divide Beyond Europe.
14:00 分子生物研究所
新科院士演講: How Xist RNA silences the X-chromosome
15:00 生物多樣性研究中心
Decoding Animal Evolutionary Novelties: From Genome Architecture to Cell Type Evolution
07/10
08:00 環境變遷研究中心
第34屆新科院士演講
10:00 原子與分子科學研究所
[2026年第34屆新科院士演講] Understanding, Controlling, and Learning from Quantum Systems
12:00 經濟研究所
Pension Reform and Asset Allocation: Evidence from Annuity Cuts to Retired Civil Servants in Taiwan
本院人文社會科學研究中心編印之《人文及社會科學集刊》第三十八卷第二期業已出版,本期共收錄6篇論文
本院經濟研究所期刊《經濟論文》第54卷第2期業已出版,本期目錄如下
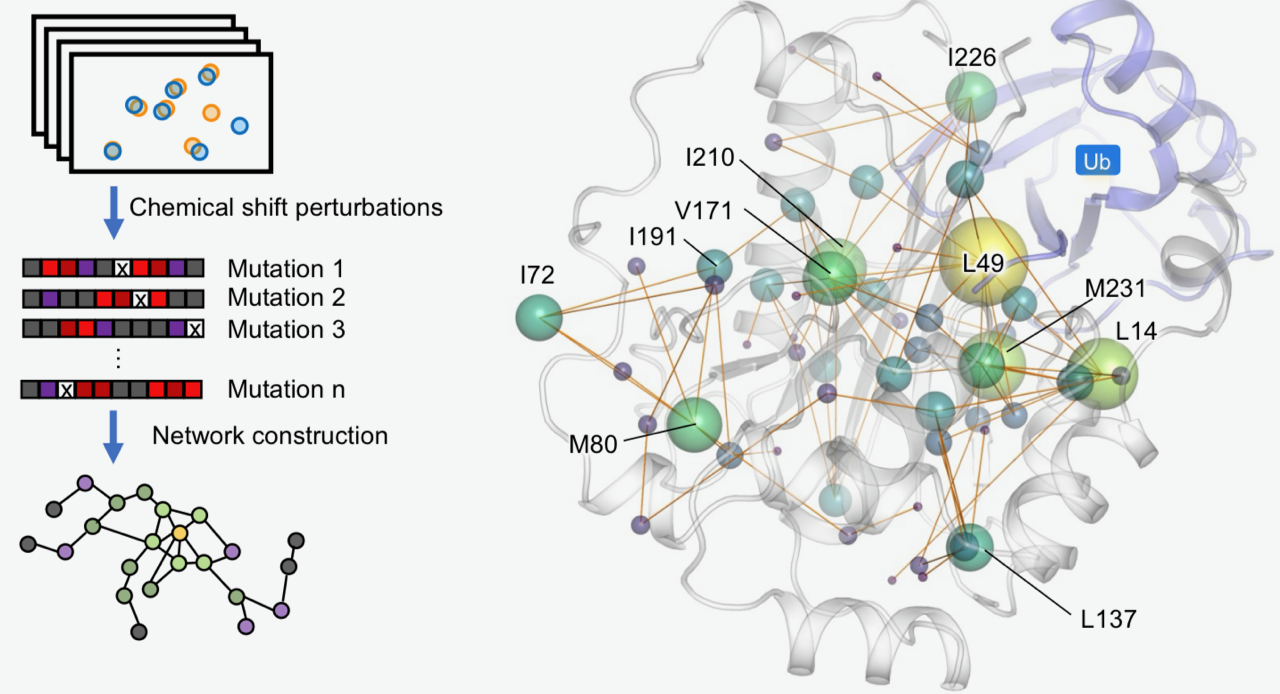
本院生化所徐尚德研究員所領導的研究團隊,結合先進甲基核磁共振(methyl NMR)、蛋白質動態分析與分子模擬技術,首度描繪與多種癌症相關的重要腫瘤抑制蛋白BAP1內部的長距離調控網絡,並系統性解析近五十個癌症相關突變的影響。
本院歐美研究所「中美關係報告」系列最新一本論文集《拜登政府中美關係專題研究》,由本院出版中心於今(2026)年五月出版。本書源自歐美所「拜登政府美中臺關係專題研究」研討會,由歐美所林正義兼任研究員主編,並力邀前總統府秘書長林碧炤教授撰寫序言,集結國內政治學與國際關係領域的學者專家,解析拜登執政時期的地緣政治角力。
微型神經探針是發展新一代腦機介面與神經植入裝置的重要工具,但其植入過程可能壓縮腦組織、損傷血管並引發出血。分生所吳玉威助研究員、研究生黃嵩文與史丹佛大學團隊攜手合作,透過結合超高靈敏度力量量測與即時活體顯微成像,直接觀察不同直徑探針插入小鼠腦部的過程。
在細胞分裂前必須先複製DNA,但同一時間,許多基因也正在被轉錄。當DNA複製機器與轉錄機器在同一段基因體上相遇,可能形成轉錄––複製衝突,造成複製叉停滯、DNA損傷,甚至造成基因體不穩定。因此,細胞如何在基因活躍轉錄區域中維持DNA複製順利進行,是基因體穩定性研究的重要課題。
過渡金屬二硫屬化物單層材料具有直接能隙、強激子響應與電場可調控的光學特性,是發展次世代光電與量子光子元件的重要平台。然而,如何兼顧大面積製造與室溫發光調控,仍是其走向實際應用的關鍵挑戰。為突破此瓶頸,本院應用科學研究中心呂宥蓉副研究員與日本東京大學化學工程系童俊智教授合作,成功將晶圓級單晶單層二維半導體(二硫化鉬),整合於原子級平坦的氮化鉿電漿子背閘極上,發展出可在室溫下操作的新型二維材料主動調變發光平台。
本院人文社會科學研究中心編印之《人文及社會科學集刊》第38卷第1期已出版,共收錄6篇論文
A型與B型流感均對全球公共衛生構成威脅,相對較常受到關注的A型流感,B型流感病毒(IBV)的研究相對較少,IBV Victoria與Yamagata兩大譜系的持續共同流行,也使疫苗預測更具挑戰性。
一般細胞在分裂多次後,染色體端粒會逐漸縮短,最後細胞會停止分裂。然而,癌細胞會利用「替代性端粒延長機制(Alternative Lengthening of Telomeres, 簡稱ALT)維持端粒長度,避免細胞老化並持續分裂。
本院近代史研究所編印之《近代中國婦女史研究》第46期業已出版
頭索動物文昌魚是脊椎動物的近親,常被用為模式生物來探討脊椎動物祖先可能具有的特徵。魯卡側殖文昌魚代表頭索動物演化上最早分支的類群,本院細生所游智凱研究員、林哲毅博士後研究學者與國際研究團隊,完成魯卡側殖文昌魚(Asymmetron lucayanum)的染色體階層基因體定序,比較基因體的結構研究發現,在頭索動物中,側殖文昌魚的基因組顯著大於過去研究的鰓口文昌魚屬(Branchiostoma)的物種。此擴張現象是由「跳躍基因」(轉座因子)的大量累積所驅動。儘管存在這些額外的DNA,其整體的基因排列順序(macro-synteny)在數億年的演化中保持得極為完整。這種穩定性很可能是因為發育過程中基因需要協同表達,因此受到選擇性限制(selective constraints)所維持的。 研究團隊亦追蹤建構身體發育藍圖的Hox基因群的演化,並辨識出脊椎動物免疫系統的古老組成要素,例如MHC和RAG基因。這項成果有助於了解無脊椎動物與脊椎動物間的重要演化轉折,為脊椎動物的基因組創新,提供極有價值的資訊。 本研究由本院關鍵突破計畫與國科會專題研究計畫支持,以及美國、韓國、中國研究機構的經費資助,本院多樣中心的高通量定序核心設施亦提供重要技術協助。論文合作者包括加州大學聖地牙哥分校Linda Holland教授,韓國中北大學Sung-Jin Cho教授,以及廣州中山大學岳家興研究員的團隊。研究成果已於2026年3月24日發表於《美國國家科學院院刊》(PNAS)。
「中研院訊」為本院重要資訊及研究成果發布平台,每雙週出刊,協助院內同仁掌握本院最新消息,也讓院外讀者更加了解本院及相關學術研究成果。
「研之有物」為本院創立之科普平臺,以深入淺出的方式報導數理、生命及人文社會領域研究成果,分享研究人員不為人知的甘苦談。歡迎進入官網吸收新知、訂閱電子報,一起探索各領域知識!








.jfif)